


SCI论文解读(18):WITER:用加权迭代回归模型拟合背景突变鉴定癌症驱动基因的统计学方法
发布单位: 广东省医学会医学科研实验室建设与管理学分会 / 阅读:2586 次 / 2021/9/6 7:49:19
 蒋琳,生物信息学方法学研究,中山大学中山医学院博士毕业,现入职广东省人民医院医学研究部。曾任中山大学中山医学院助理研究员,香港大学医学院基因组研究中心研究助理。以人类疾病遗传定位计算方法创新为主要研究方向,在癌症驱动基因定位和复杂表型原发组织定位方面提出了高效能的新方法,不仅丰富了现代遗传学定位方法的理论,也为实例研究节省了大量的资金和时间。发表SCI论文5篇,其中以第一位共一作者在Genome Biology、Nucleic Acids Research期刊发表方法学创新论文2篇。
蒋琳,生物信息学方法学研究,中山大学中山医学院博士毕业,现入职广东省人民医院医学研究部。曾任中山大学中山医学院助理研究员,香港大学医学院基因组研究中心研究助理。以人类疾病遗传定位计算方法创新为主要研究方向,在癌症驱动基因定位和复杂表型原发组织定位方面提出了高效能的新方法,不仅丰富了现代遗传学定位方法的理论,也为实例研究节省了大量的资金和时间。发表SCI论文5篇,其中以第一位共一作者在Genome Biology、Nucleic Acids Research期刊发表方法学创新论文2篇。

1. 引言
体细胞中基因组失常是引起癌症的主要驱动因素,根据癌组织中基因体细胞突变数鉴定癌症驱动变异或基因并探测全面的癌症驱动基因图谱,对有效诊断和治疗癌症至关重要。然而,癌症有很强的异质性,而且大部分癌症驱动基因是温和的且影响不强烈,加上大量中性伴随突变的干扰,使得探测驱动基因很困难。本研究提出了一种全新的对变异位点功能加权的探测驱动基因统计方法,加权迭代截零负二项回归模型 (免费公共平台下载KGGSeq, http://grass.cgs.hku.hk/limx/kggseq 或WITER, http://grass.cgs.hku.hk/limx/witer)。
2. 方法简介
WITER突破现有方法常用泊松分布模型拟合背景突变的模式,创新性地引入负二项分布,并通过反复探索和改进,首次用截零负二项回归模型估算癌细胞非同义突变。进一步,WITER将变异位点对基因功能的影响直接整合到位点权值中,达到放大或缩小体细胞突变数的目的,使体细胞基因突变负荷能充分综合变异对基因功能的影响。此外,WITER采用借用独立参照样本的策略使小样本也能构建稳定的回归模型,即便只有30左右TCGA小样本集也能探测到显著驱动基因。WITER通过有效地拟合背景基因突变数分布,不仅显著提升了鉴定效能、定位更准,还解决了困扰多年的中、小样本量效能低下的难题。方法主体结构如图 1所示
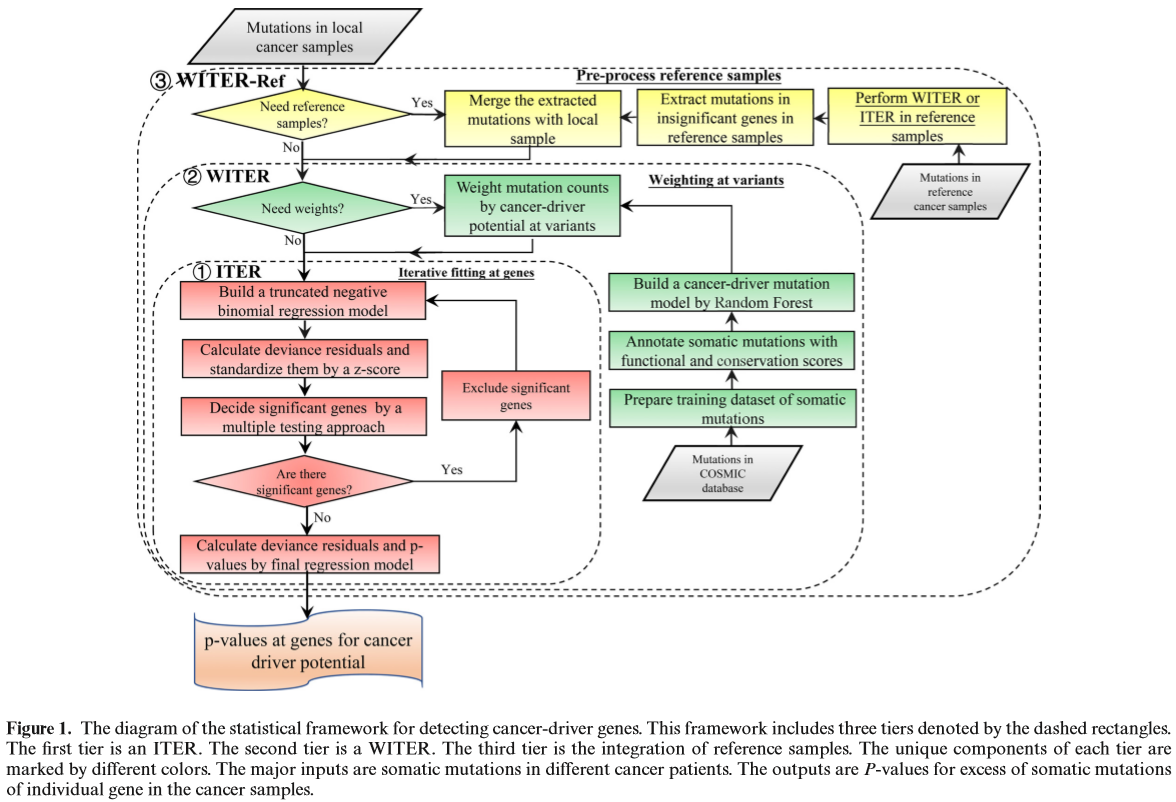
WITER的强大效能源自于4大技术创新:首次截零负二项回归模型拟合背景突变、通过迭代纯化背景、对基因突变数加权增加效能、借用参照样本稳定模型,使WITER能突破样本量局限构建了先进稳定的理论模型。
3. 对比验证及应用
WITER与4种有代表性的同类方法做对比验证分析,WITER几乎都能探测到最多的显著癌症驱动基因和CGC驱动基因,同时避免了统计显著紧缩现象,能挽回最多的被其它方法漏检的驱动基因,且速度最快,详见图2 。
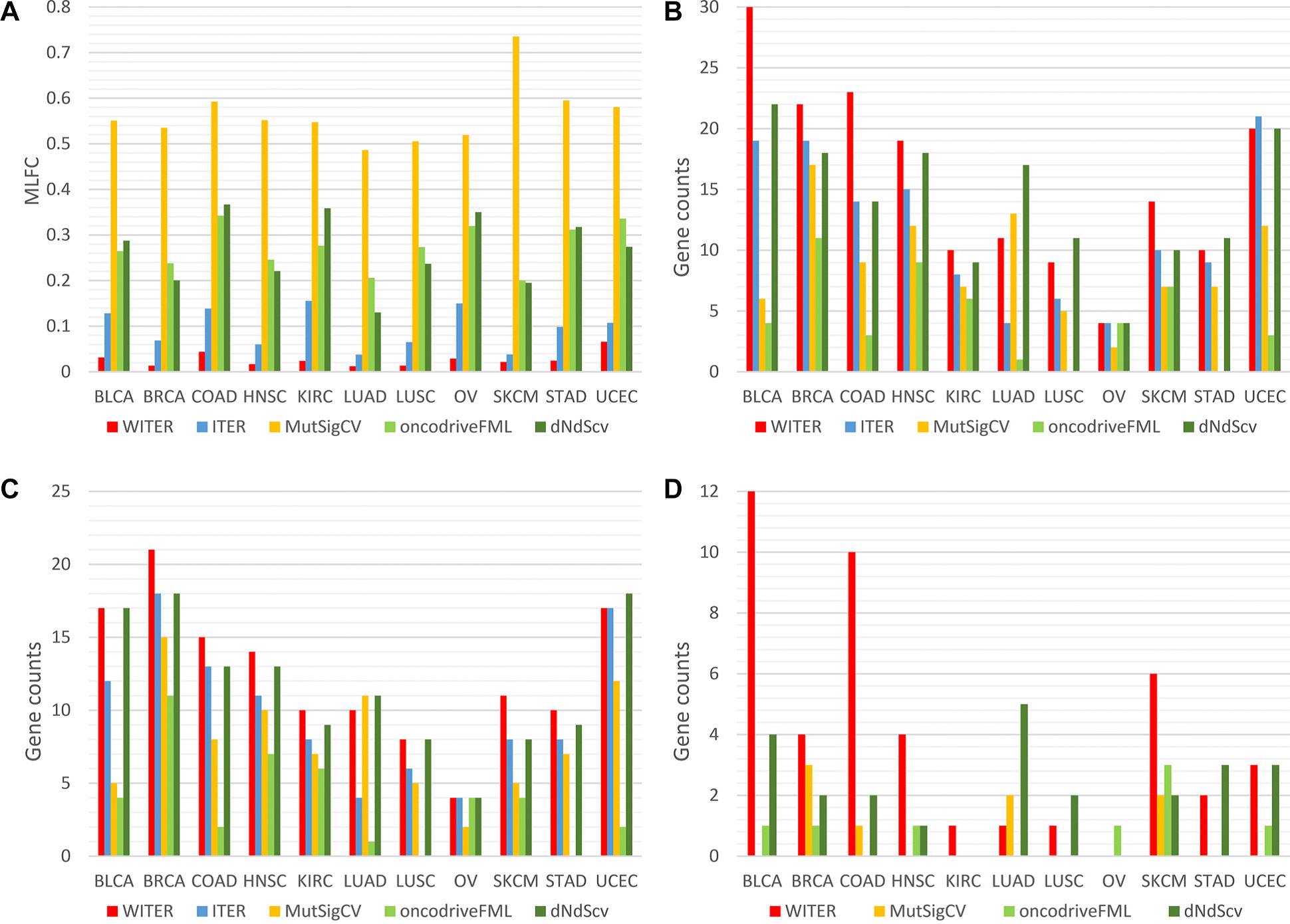
Figure 2. Performance comparison of differentmethods for detecting cancer drivermutation in 11 cancers. (A) TheMLFCof four methods; (B) the number of significant genes; (C) cancer consensus significant genes; (D) the number of unique significant genes. The P-values less than a cutoff according to FDR q 0.05 are excluded. The full names of cancers are in Supplementary Table S1.
将WITER应用于TCGA为主的癌症样本,为26种癌症类型探测到了229个驱动基因(图3),其中23种癌症类型的独有显著基因100多个。In-silico(文献检索验证)验证了显著基因中有78%是已知驱动基因或与已知驱动基因有关,而很多其它显著基因也有依据推断可能是未被发现的新驱动基因。因此,生成的26种癌症类型潜在全局图谱为诊断和治疗癌症提供了清晰明确的潜在标记。

Figure 3. Circos plot displays 178 significant genes in 26 cancers. Notes: The innermost ring denotes dendrogramof genes. The next ring contains significant genes (marked in red) of corresponding cancers. It is followed by a ring of cancers inwhich the genes are significant. The outmost ring contains gene symbols. The full names of cancers are in Supplementary Table S1.
4. 总结
该研究不仅为受中小量样本局限的基因组变异数分布提出了先进的理论模型,还研发了一个用于探测潜在癌症驱动基因的有效工具,也为基于体细胞突变绘制了一幅图多肿瘤驱动基因全局图谱。
原文链接:
https://academic.oup.com/nar/article/47/16/e96/5530310 (Nucleic Acids Research, IF = 11.147)
特别鸣谢:
本文转自:GD省医公共实验室公众号

欢迎投稿:MLCMGD@163.com